• Sources
of Cancer Inducing Mutations (click picture)
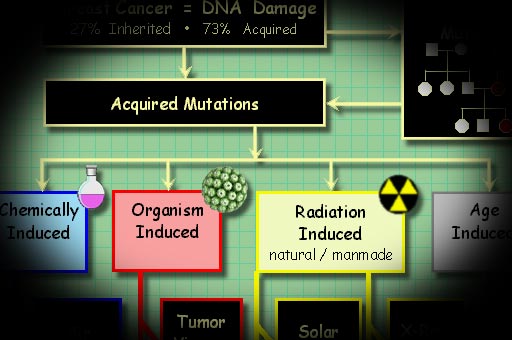
Cancer
is a disease, first of all, of cellular mutations.
The sources of these mutations
are charted in the figure above.
Many researchers believe that five
to ten critical mutations must accumulate to
create a cancer cell line, also known as a clone
line.
Cancer can be viewed as an accumulation of genetic
incidents, that begin with DNA damage, mutation
or silencing. These precancerous cells undergo selection for
specific growth advantage that ultimately
leads to metastasis. These mutations may proceed at a higher rate after the clone line
has multiplied due to a breakdown in cell cycle
quality control checkpoints, much like a runaway
train jumping the tracks. Just as antibiotics kill bacteria,
chemotherapy kills cancer
cells. Then, just as in antibiotic resistance, chemotherapy kills cancer cells except those that have the ability to grow
in the more difficult circumstances.
The resistant tumor cells are then selected and
continue to grow and proliferate in a Darwinian survival
of the fittest fashion. New biologic therapies using monoclonal antibodies may
bypass this process of
cellular evolution. Cells that grow and cycle rapidly are statistically more vulnerable to the mutations that cause cancer. These include skin cells and cells that line the digestive and reproductive tracts. These kinds of cell grow and turn over rapidly - fast growth and replacement means more chances for things to go wrong.
There
are two overarching categories of genes implicated in cancer, tumor suppressors and oncogenes.
An example of a tumor suppressor
genes is the famous p53 cell-cycle checkpoint gene you may have heard about. Tumor suppressor genes can
be likened
to
the brake pedals in a car, slowing cells from dividing too rapidly. The second category is oncogenes,
or growth genes, like those activated by steroids.
When
oncogenes turn on, cells multiply.
Oncogenes are like the accelerator pedal
of a car. It is a disaster when the accelerator
sticks and you can't turn it off.
Cells begin as primal undifferentiated
cells called stem cells. Stem cells are general purpose blanks that can become any specific type the body needs. On receiving certain chemical signals called cytokines,
stem cells differentiate or morph into
the specific tissue type required. A well understood example of this are circulating cells - red cells, white cells and platelets. They are manufactured in the bone marrow, where they are exposed to specific cytokines that tell them what kind of a cell to become.
Mature cells of specific
tissue types may
revert to more primal
forms when
signaled. In cancer, some fully differentiated cells
appear to "back up" to their more primitive
states, and then grow out of control.
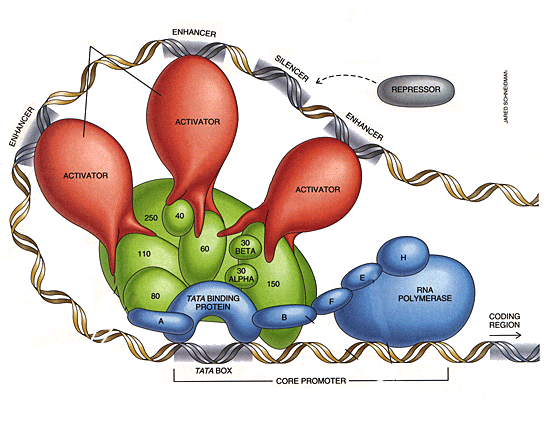
Genes consist of sequences of DNA
bases chosen from the
four letter set: C,T,A or G. Genes are the literal blueprint for what protein the body should make. This is done in two steps, transcription and translation. In transcription the DNA is scanned by a special protein complex called RNA Polymerase and converted to an RNA transcript that holds the instructions for making a protein. A protein can be a structural protein like collagen, or a cell surface receptor protein that transmits signals, or a housekeeping protein for metabolism. The transcription of DNA to RNA is done by RNA polymerase, a complex protein assembly with several working parts. It has upstream sensors that determine how actively to transcribe DNA. The BRACA1 gene is part of this transcription apparatus, the copying machine if you will. When BRACA1 is broken, sequences that are transcribed/copied can mutate. The figure below shows the RNA Polymerase enzyme complex. It scoots along the DNA strand until it hits a stop signal. The animation below shows it in action.
With transcription complete, a messenger RNA transcript is reading to be translated into protein. Groups of three DNA letters are read, three
at a time, and these three letters specify which amino acid will be ultimately be chosen for the protein sequence. There is
an alphabet of twenty amino acids that make
all proteins. The translation process is animated below:
When the DNA
is duplicated
during
cell growth, an error may then occur according to the table at the top of this page. Lexically,
there are three categories of errors in DNA, deletion,
substitution,
and insertion. Genes, coded in the 4 symbols of DNA, can be thought of as strings of letters
that must satisfy parsing rules and grammar just like any language.
In
the first error category of deletion mutations,
one or more of the
letters
is missing.
DNA bases are read in groups of three to produce
protein,
one amino acid at a time. DNA bases taken in
groups of three letters are called codons.
Deletion of one or more of these letters throw
off the entire protein coding sequence for that
gene. This results in nonsense downstream of the
mutation. DNA has
some built in error correction, due
to the double helix. This bears similarity to a
Hamming error correcting code in computer science.
Nucleotide bases pair across the helix with their
complementary
base.
Cytosine, C, always pairs with guanine G,
by hydrogen bonding across the rung of the DNA
ladder.
Similarly adenine, A, pairs with thymine, T.
DNA repair
enzymes
such
as
DNA polymerase and DNA ligase correct missing
or
incorrect
bases
provided that the complementary base
on the other strand is correct and provided
a carcinogenic substitution has not occurred. Individuals
whose repair enzymes are defective, such as those
with
XP, Xeroderma pigmentosum, may develop tumors spontaneously
on exposure to sunlight. The UV component of sunlight is damaging to DNA. In normal individuals, this
is repaired by DNA polymerase. Asymmetry of repair
is the rule of the day. The leading and lagging
strands of DNA are copied and
repaired
with different enzymes and topological functions
resulting in a different likelihood of
successful replication for each side of the DNA
helix.
The
second category of mutation is substitution,
where one
letter is substituted for another. The seriousness
of this error depends on whether it is the
first, second or third base in the codon. Single
letter
substitutions frequently result in a codon
that specifies the same amino acid. There are 64
codons, because there are 64 ways of ordering a
triple of C, T, A, or G. But nature has caused
these 64 primitive instructions to map to only
20 amino acids, so the 64 codons are not unique.
As
in a
digital
numbering
system, the leading letter of
the codon
is most
significant.
Two different codons can code for the same amino
acid, thus a substitution mutation will frequently
make no change in the gene product. Often the protein
will function properly if a similar amino
acid is
substituted.
This
is not always the case. Substitution mutations
are also called point mutations. Sickle cell anemia
is caused by a single point mutation in the gene that codes for hemoglobin. Hemoglobin is a globular
protein that serves to transport an oxygen diamond
in the setting of a porphyrin ring. Sickle cell
hemoglobin will polymerize with
itself under conditions of low oxygen tension.
Sickle cell and cancer are not related, but the
mechanisms, history and legacy of mutation are
related.
The
third category of mutation is an insertion mutation.
In this mutation, one or more letters are inserted
into a coding sequence. If three letters or a multiple
of three letters are inserted, the protein may
still function properly. If a non
multiple of three letters is inserted, nonsense
will again result for all codons downstream of
the insertion mutation. Viruses can cause insertion
mutations, since their DNA is incorporated into
the host cell's DNA. An interactive gene sorting illustration
of the genetic wreckage accumulated by the BRCA1 breast
cancer
gene
is shown
below.
There
are two kinds of DNA sequence, those stretches
that code for protein product, called exons,
and those
that do not code for protein, called introns. The
latter are sometimes called, "junk DNA",
because no function is known for this genetic material.
About 96.4% of the human genome is "junk DNA".
This is quite remarkable. Of that 50% consists of 742 repeating idioms shown below, some of which are quite similar to each other. You may click on the image to see the full size poster.
Examples
of these three kinds of mutations can be seen
in the BRACA1 gene rollover graphic below. Placing
your mouse
over the graphic, causes the components of
BRACA1 to be sorted. A common repeated intron is
called an ALU repeat. The ALU repeats in BRACA1
can be seen to be profoundly frame shifted and
mutated.
Fortunately these repeats do not code for gene
product, but they portend the mutations that do occur
in the exonic regions, that do contribute to
breast cancer.
Deletion
and insertion mutations are also called frame shift
mutations, because they alter the codon reading
frame and produce nonsense. Mutations rarely
result in a gain of function. Mathematically,
loss of function
mutations are much more likely.
Mutations
are not the only way that important genes can be
damaged or silenced. Genes have switches called promoter
and repressor regions. When certain proteins bind
to the promoters or repressors the amount of protein
produced from
the
gene can
be turned up, turned down, or turned off. Another
mechanism for silencing genes is methylation. In
gene methylation,
a cytosine base in the gene's promoter region
is tagged with a methyl group. Like protein binding
to promoters, methylation is an important natural
mechanism for gene regulation. But when a tumor suppressor gene is silenced by methylation, serious
trouble may result.
Most
cancer can ultimately be traced to DNA damage, mutation
or gene silencing by methylation. The source of unwanted
methylation is unknown. The enzyme that enables
methylation is the enzyme methyl transferase. Thus
tracking down sources of methyl transferase, for
example in viruses that are known to cause cancer,
would be an interesting line of investigation.
Looking at mutations that affect the expression of
methyl transferase would be another.
Mutations
can be inherited from a previous generation, if
the mutation appeared in the germ-line. Conversely they can
be during the lifetime of the cell. The same
is true of methylation.
Cancer
is also a disease of age. When DNA is replicated,
there must be extra length at the ends of the strands
to allow the duplicating enzyme to stay on the tracks,
so to speak. These extra lengths are called telomeres
and are maintained by an enzyme called telomerase.
Telomerase is a reverse transcriptase. When telomerase is absent, the
ends
of the chromosomes
fray. Important gene products coded by these fraying
ends then fail to be made and the deterioration
of aging ensues.
--
SKY image kindly provided by Dr. Jeff Sawyer, Arkansas
Children's Hospital
The
human body consists of 37 trillion cells divided into several
organ systems and tissue types. These specialized
tissues consists of groups of similar cells, e.g. liver
cells, skin cells, nerve cells etc. Each cell contains
its own copy of the human genome, the chromosomes that
carry the instructions for how to make a complete human
being. Chromosomes are made of DNA and 96.4% of the DNA
does not carry instructions for anything useful. Only
3.6% of our DNA contains useful genetic information.
At the end of the cell cycle the genome consists of 46
chromosomes, 23 provided by each parent. Each chromosome
contains a few thousand genes separated by long deserts
of non-coding DNA. In the human genome there are 20,376
confirmed genes. In differentiated, mature tissues,
most genes are turned off, except for those necessary for housekeeping and essential cell
function. Genes are the instructions to make a given
protein. Proteins can be enzymes, which enable chemical
reactions. Proteins can be structural materials such
as tubulin which give cells internal form. Proteins affect
shape as in collagen which
connects cells to each other.
In
reproductive cancers, hormone sensitive cells derived
from epithelial cells grow rapidly in an uncontrolled
manner forming a mass of unwanted tissue called a tumor.
Cancer cells express proteins that dissolve collagen
and enable them to break away and travel to other parts
of the body. These proteins are called serine proteases,
or MMP's. They are similar to digestive enzymes.
When
certain growth genes and cell regulation genes such as
p53 are broken from mutation, the cell will divide before
its DNA has passed certain "quality control" checkpoints.
When this occurs, frayed ends of the chromosomes in the
cell can rearrange into new configurations such as the
one shown above. Pieces of one chromosome can fuse to
another in a process known as translocation.
A
technique called Spectral
Karyotyping is informative about translocations that
occur in a variety of cancers.
Rapidly
cycling hormone sensitive tissues such as breast, ovary
and prostate, are much more likely to contain mutated
DNA. Hormones themselves do not cause cancer, but hormone
induced DNA transcription and cell cycling increases
the probability of mutation.
Characteristics
of hereditary breast cancer:
Multiple
cases of early onset (< age 45) cancer
Associated
ovarian and/or colon malignancies
Vertical
transmission (maternal or paternal lines)
Bilateral
breast cancer
•
Five to Ten Mutations = A "Full House"
Each
of the 37 trillion cells in the body carries
its own unique configuration of mutations.
Most
of the time, these mutations are harmless
because they occur in noncritical sections
of DNA, or are corrected by the DNA repair
enzymes before replication occurs.
Sometimes
therapy can help and hurt at the same time.
For example, chemotherapy
kills cells that are highly mutated, at the
cost of increasing the mutation level of normal
cells.
Mutations enable:
immortalization
uncontrolled growth
metastasis
angiogenesis
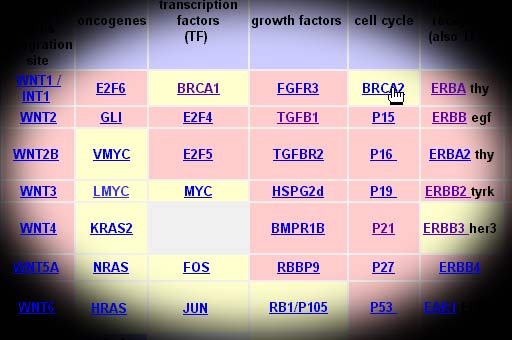
Oncogenes
found in biopsy specimens of breast carcinoma
cells include:
Immortalization
is considered to be one important transformation
in a cancer cell line. Without immortalization
the cell can only multiply 50 times before the
chromosomes ends. How
many cells can be made in 50 divisions? How
much would a tumor made of such cells weigh?
200
Breast Cancer Genes:
Grouped By Function
Tumor
Supressors And Cell Growth (Oncogenes)
•
The BRCA1 Breast Cancer Gene
In
1990, through genetic linkage studies, a gene(BRCA1)
was localized on chromosome 17 at band q21
that predisposes to a significant proportion
of early-onset breast cancer. Later studies
found BRCA1 increased susceptibility for ovarian
cancer as well as breast cancer. A
study of 214 families linked to 17q21 the cumulative
risk for breast cancer associated with
BRAC1 to be about 59% by age 50 and 87 % by
age 70.
Another
gene (BRCA2) has been implicated in hereditary
breast cancer. This gene has been localized
to a region on chromosome 13q12-q13.
Several
families linked to BRCA2 show breast cancer
in women as well as men, whereas no breast
cancers in men have yet been observed in families
linked to BRCA1.
The
p53 gene also has been linked to breast cancer.
This gene is a tumor suppressor gene which
normally acts as a transcription factor that
regulates the expression of other genes and
has been mapped to chromosome 17p13.
A
variety of different types of mutations in
this gene have been found.
Investigators
are now showing cautious optimism for using
p53 as a prognostic tool; the over expression
of p53 correlates with a poor prognosis in
node-negative breast cancer.
The OMIM
morbidity map shows this and other cancer
genes. Genes implicated in the cancer process
are called oncogenes.
A
= aqua C = black G = gold T = Teal
The
gene called BRCA1 actually contains instructions
for four different proteins, IFP35, BRCA1,
RHO7 and IFB35. These instructions are called
exons. If you place your mouse on the image
you can see these exons at the very top in
the sorted version. The next region down with
a regular pattern consists of non-coding DNA
called ALU repeats. These make up 35% of the
BRCA1 gene. These are the regions in the middle.
The smeared appearance is due to deletion
and point mutations that have occurred over
the entire history of this gene.
•
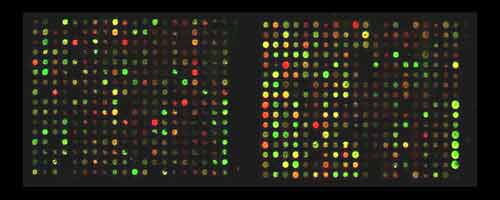
Gene Chips
Characterizing Mutations In Breast Cancer
Breast
cancers that were formerly reported as being
the same, are being screened using gene chip
technology to create a personal
portrait of an individual's specific cancer.
These arrays may used in the future to customize
therapeutic regimens based on the information
on the specific cancer clone. In this way,
patients can be treated according to the specific
pathways that are active in their specific
tumors. Data on how they respond to these
treatments can be incorporated into treatment
databases to further improve therapy.
Groups
of genes implicated in carcinogenesis can
be clustered into groups
that characterize the activity of entire pathways
that are up or down regulated in the disease
state.
To
explore a map of breast cancer gene expression
click here.
Gene sequencing is a powerful
tools in the fight against cancer.
It is now possible to compare gene expression
in a patient's tumor with those in databases
and in cell lines. By using combinatorial
chemotherapy techniques on cell lines, we
can know in advance which
chemotherapy drugs have the best chance to attack the tumor effectively.
The
links below provide more information on genes
involved in cancer.
We
now live in the era of a sequenced genome, gene
chips, monoclonal antibodies, fast computers and
the internet. With this armada of powerful weapons
a new approach is emerging to solving the cancer
riddle. Applying computers to the cancer problem
is yielding unexpected avenues of progress. One
such avenue is called knowledge
mapping. Two knowledge maps that would yield
great insight are the oncogene knowledge map (200+
maps) and the gene expression knowledge map (1763
maps). The completion of these first maps will
greatly increase our understanding of cancer and
will enable simulation of cellular processes.
In addition to knowledge mapping there are interspecies
comparative genomics projects from the field of gene ontology.
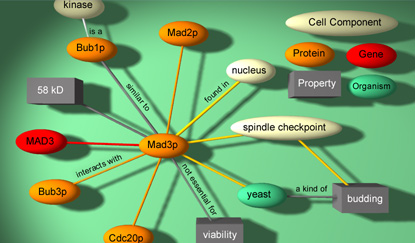
• Knowledge
Mapping
New approaches are
being explored in the field of bioinformatics.
These approaches are using data-mining techniques
developed in disciplines that are far removed
from traditional oncology. A demonstration
of these techniques is here.
Two exciting technologies that are certain to have a significant impact on cancer diagnosis and treatment are: